STUDIES OF LOCAL STRUCTURE IN POLYMERS
USING X-RAY SCATTERING
M.J. WINOKUR
Department of Physics
University of Wisconsin
Madison WI 53706
INTRODUCTION
The study of the intrachain and interchain structure in polymeric materials has been an exceedingly important research topic and techniques which can resolve these attributes remain one of the central research pillars used to characterize systems composed of these chain-like molecules. The enormous anisotropy intrinsic to the polymer chain structure is the defining materials property and, as a result, one is interested in structure and structural phase behavior at a large number of length scales. For investigations at the largest microscopic distances, ranging from 50 to 10,000 Å, small-angle neutron and x-ray scattering and light scattering techniques[1,2] are often employed and these studies can yield a vast amount of detailed data. At shorter length-scales wide-angle x-ray, neutron and electron diffraction techniques all provide complementary information about crystalline, semi-crystalline and amorphous hosts[3,4,5].
In large part the most quantitatively accessible features are those generated by
crystalline and semi-crystalline materials. In this case the elastic Bragg
scattering, at wave vectors q > 0.1 Å-1 (as obtained from ![]() where
where ![]() is the scatterer wavelength and
is the scatterer wavelength and
![]() is the angle between the incident beam and the scattered beam),
provides direct information about the placement of the polymer chains within a
periodic unit cell. Since the chain chemical architecture is generally a known
attribute, it is often possible to ascertain the average geometric construction
of the polymer host. Still it is important to emphasize that, even in the
best-case scenario, this analysis is most sensitive to the interchain packing of
the polymer chains and, in general, the unit cell contains a large amount of
static and and dynamic disorder. Hence all of the Bragg scattering undergoes a
rapid exponential falloff with increasing q so that by 4 Å-1 or so this
scattering signal is no longer resolvable. Moreover most crystalline polymers
contain a considerable fraction of amorphous material which generates a
secondary diffuse scattering background. In semi-crystalline hosts the
proportion of diffuse scattering signal is increased. Finally I note that many
polymeric materials, including those in the melt state, exhibit only diffuse
scattering signatures and thus are essentially amorphous or liquid-like.
Obviously, in these cases, there can be no analysis of the Bragg scattering yet
an understanding the atomic-length-scale structural organization is still
desirable.
is the angle between the incident beam and the scattered beam),
provides direct information about the placement of the polymer chains within a
periodic unit cell. Since the chain chemical architecture is generally a known
attribute, it is often possible to ascertain the average geometric construction
of the polymer host. Still it is important to emphasize that, even in the
best-case scenario, this analysis is most sensitive to the interchain packing of
the polymer chains and, in general, the unit cell contains a large amount of
static and and dynamic disorder. Hence all of the Bragg scattering undergoes a
rapid exponential falloff with increasing q so that by 4 Å-1 or so this
scattering signal is no longer resolvable. Moreover most crystalline polymers
contain a considerable fraction of amorphous material which generates a
secondary diffuse scattering background. In semi-crystalline hosts the
proportion of diffuse scattering signal is increased. Finally I note that many
polymeric materials, including those in the melt state, exhibit only diffuse
scattering signatures and thus are essentially amorphous or liquid-like.
Obviously, in these cases, there can be no analysis of the Bragg scattering yet
an understanding the atomic-length-scale structural organization is still
desirable.
This diffuse scattering signal also contains a superposition of information concerning both the local interchain and intrachain structure of the polymer and, in many instances, it is this local structure which is the most relevant feature for understanding the physical properties of the polymer host. The recent and rapid development of molecular level materials engineering and new light sources[6] in combination with computer generated atomistic simulations of polymeric materials[7,8,9] has initiated a resurgence of interest in direct techniques which can adequately resolve the local chain structure. Simulations which do not accurately describe the local structure may not be expected to faithfully reproduce physical properties at larger length scales. Moreover there are a number of novel materials which are extraordinarily sensitive to the nature of the local structure. In conducting polymer hosts electronic charge transport requires motion of charge both along the backbone and between chains to create a three-dimensional conducting matrix. In these materials subtle variations in both the intra- and inter- chain organization can generate profound changes in the measured transport properties[10,11,12].
To adequately reconstruct structure at length scales ranging from 1 to 10 Å it
is often possible to employ radial- (or pair-) distribution-function analysis
(RDF or PDF respectively) techniques. PDF analysis can be successfully used to
elucidate structure in a variety of structural ``settings'' ranging from
crystalline to amorphous[13]. These techniques can even be exploited in polymer
samples containing appreciable two phase mixtures of crystalline and
amorphous components[14]. A number of excellent reviews of radial (or pair)
distribution function analysis have appeared in the literature [3,15,16] and some these
are even specific to polymers[17].
The overall goals in the text that follows are to
briefly review a few of the most recent PDF studies of polymeric materials, to
highlight the unique attributes of PDF analysis in polymers, and finally, to
describe in modest detail some of the specialized difficulties and solutions
for obtaining quantitative analysis of the local chain structure when using
refinement of x-ray scattering data.
BASIC THEORY
PDF studies require data analysis and reduction techniques which are considerably different than those typically employed for analyses restricted to only the Bragg-like scattering features. In general it is necessary to acquire all scattering data, from moderately low to relatively high wave vector (or q), correct the experimental data for a variety of systematic effects[15] (e.g., geometry, absorption, multiple scattering[18], x-ray Compton scattering[19], backgrounds, etc.) and then perform a properly normalized Fourier transform. In theory this new spectrum contains a weighted distributional average over all atomic pairs. Because of the fundamental complexity in deciphering the superposition of all these distributions it is necessary to choose host system carefully so as to take best advantage of the resultant data. For polymeric materials it is desirable to choose model compounds containing a very limited number of monomer base units [e.g., polyethylene or poly(propylene)] or anomalous scattering scattering centers (e.g., large-Z cations or anion species in polyelectrolytes). In the latter case it then becomes necessary to employ differential anomalous scattering[20] methods.
A common starting point for this analysis is the Debye formula
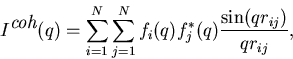 |
(1) |
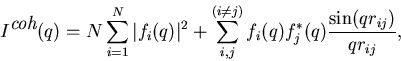 |
(2) |
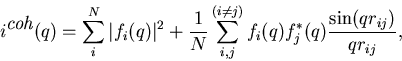 |
(3) |
![\begin{displaymath}
H(q) = \frac{[i^{\mbox{\it coh}}- \langle f^2 \rangle]} {\la...
...\mbox c}_{i'} {\mbox c}_{j'} f_{i'}(q)
f_{j'}^*(q) e_{i'j'}(q) \end{displaymath}](img8.gif) |
(4) |
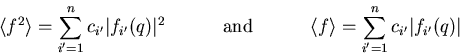 |
(5) |
![]()
| (6) |
POLYMER SPECIFIC ISSUES
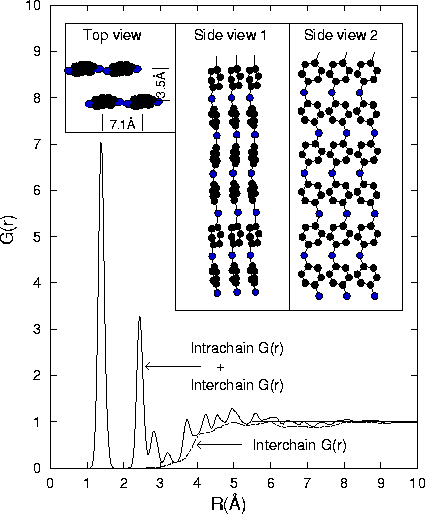
The central goal is to develop quantitative methods for resolving both the interchain and intrachain structure in polymeric compounds using either x-ray or neutron based scattering methods. Because there exists an enormous difference between these two structural components, polymer PDF profiles manifest a rather unique line profile. In the case of amorphous polyaniline[23], a relatively rigid rod-like polymer given by
 |
![\begin{figure}
\begin{center}
\begin{center}
\includegraphics [height=3.5in]{fig...
....eps}
\end{center} \vspace*{-.1in}
\end{center} \vspace*{-.20in} \end{figure}](img21.gif) |
In all situations scattering data out to very high momentum transfer is a
prerequisite for a comprehensive analysis. For neutrons, with their q
independent atom scattering factors, this does not present an undue
complication. X-rays are more problematic. By 20 Å-1 the carbon
structure function has diminished by almost 95% and is now superimposed on a
Compton scattering background almost nine times larger. A typical intensity
spectrum highlighting this loss of scattering intensity is shown in Fig. 3.
Moreover the intrachain scattering is only a small fraction of the total
coherent scattering signal. Ideally a large aperture detector (to increase the
effective count rate) combined with exceptional energy resolution (to fully
isolate the coherent scattering) would be best. The limited energy resolution
of current detector technologies allows for only a partial separation although,
as the inset of Fig. 3 shows, the Compton profile can be adequately resolved at
higher q-values. The actual shapes of the both the elastic and Compton
scattering profiles can be ascertained so that one only need vary the
intensities of the two constituents. By fitting all energy dispersive profiles
at the higher q-values the ratio of these two components can be experimentally
deduced. To reduce the ![]() statistical noise still present in the
extracted Compton profiles this data is typically passed through a low-pass
Fourier filter[25]. At some intermediate q value the coherent
scattering profile
must then be suitably spliced to the existing low-q experimental data. In
this way quantitative x-ray data is obtained out to reasonably high q values.
For x-ray studies, as compared to those using neutrons, there is an addition
advantage in that the hydrogen atom scattering is essentially negligible at q
values beyond 10 Å-1 so that the structure function is dominated by
only the major skeletal atomic constituents.
statistical noise still present in the
extracted Compton profiles this data is typically passed through a low-pass
Fourier filter[25]. At some intermediate q value the coherent
scattering profile
must then be suitably spliced to the existing low-q experimental data. In
this way quantitative x-ray data is obtained out to reasonably high q values.
For x-ray studies, as compared to those using neutrons, there is an addition
advantage in that the hydrogen atom scattering is essentially negligible at q
values beyond 10 Å-1 so that the structure function is dominated by
only the major skeletal atomic constituents.
![\begin{figure}
\begin{center}
\begin{center}
\includegraphics [height=4.5in]{fi...
...fig3.eps}
\end{center} \vspace*{-.1in}\end{center} \vspace*{-.10in}\end{figure}](img23.gif) |
An absolute measure of the coherent/Compton ratio has an additional benefit which is of some consequence. Often the sample geometry (i.e., reflection, transmission) in combination with absorption effects requires pronounced corrections of the profile shape function. These corrections can be very sensitive to sample alignment and measurements of the absorption coefficient. Moreover polymer samples are often macroscopically inhomogeneous; a property which further impacts these corrections. By obtaining this direct measurement of the relative coherent and Compton fractions one can, after appropriately scaling the experimental ratio to match the theoretical curve, immediately execute a Krough-Moe normalization[26] of the data without resorting to many of these aforementioned corrections.
Even after a rigorous attempt is made to apply all of the generally accepted curve correction schemes[15], these extended q profiles can still display small systematic deviations which both hamper direct refinements of the structure function and create unphysical artifacts at low r. Although a thorough investigation establishing the true origin of these discrepancies has not been attempted it may well be that the derivations of the theoretical atomic scattering curves themselves are not sufficiently accurate. Despite these limitations there are a few ad hoc procedures which can ``correct'' the data without imparting significant new systemic errors in the spectral regions of interest. One correction scheme is to identify nodes of the structure function (where H(q)=0) from a representative model and perform a series of Krough-Moe normalizations. This information is then used to generate a correction curve to the nominal full-range normalization procedure. To supplement this approach it is also possible to effect a low r (typically at 1.2 Å and less) correction of G(r) by simply using the known profile as generated by a representative model and replace the low r portion of G(r). This is then Fourier back transformed to yield an additionally modified H(q). Figure 4 depicts the sequential variations in the structure function as each indicated modification is implemented.
![\begin{figure}
\begin{center}
\begin{center}
\includegraphics [height=4.5in]{fig...
....eps}
\end{center} \vspace*{-.1in}
\end{center} \vspace*{-.20in} \end{figure}](img24.gif) |
There is also the ubiquitous problem of artifacts generated in the Fourier
transform procedure itself. As Fig. 2 clearly demonstrates, significant
oscillatory behavior exists in the structure function well beyond the highest
q-values accessed. As such, a simple transform of data is expected to produce
a variety of truncation effects including a broadening of the G(r) peaks and
the so-called ``ringing'' artifacts (which arise from the Fourier transform of a
step function). One seemingly effective solution, proposed by Mitchell and
Lovell[27], is to execute a `sampled transform' in which a
series of fast Fourier transforms are overlaid using the same H(q) data set
having fewer and fewer high q data points. In the way the ringing occurs at
different frequencies so that, on average, this undesirable feature is
minimized. Moreover, the low q data, which typically has less statistical
error, is weighted more heavily. The Fig. 4 inset shows a typical real space
profile that is obtained after this scheme has been employed. Other procedures,
such as artificial damping and/or extension of the H(q)[28], have also been
applied with varying degrees of success.
EXPERIMENTAL DATA AND MODELING
Ultimately the experimentally derived correlation functions and structure functions need to be rigorously compared against a physically realistic model. Since the actual chemical organization is predetermined, this gives a well-defined starting point for undertaking these comparisons. Still there is a formidable number of pair correlations even in the most basic of polymer model compounds [e.g., polyethylene or poly(tetrafluoroethylene)]. In the simplest setting, only the nearest-neighbor, next nearest-neighbor and greater distances are used assuming a nearly isotropic root-mean-square (rms) distribution of the atomic positions and a fixed chain geometry[28]. In this case the eij(q) term in Eqn. 5 is approximated by
| (7) |
Detailed knowledge of the local pair, bond-angle, and torsion-angle potentials
enables a far more sophisticated modeling approach. Since these atomistic models
contain all atom locations, including disorder, the need for employing
the exponential term in Eqn. 5 becomes unnecessary for evaluating q H(q).
In the case of
hydrocarbons and fluorocarbons a number of full-fledged molecular simulations, using either
molecular dynamics or molecular mechanics, have been performed and the
computationally generated curve profiles compared to
experimental data[29,30,31].
As the
recent work of Londono et al.[31]
in Fig. 5 shows there is relatively good
agreement between x-ray experiments and theory for a ``simple'' hydrocarbon
system, in this case for isotactic poly(propylene) in the melt. Neutron based
experiments, which allow for much higher q ranges, of polyethylene and
poly(tetrafluoroethylene) melts have also demonstrated the utility of this
approach.
![\begin{figure}
\begin{center}
\begin{center}
\includegraphics [height=3.5in]{fig...
....eps}
\end{center} \vspace*{-.1in}
\end{center} \vspace*{-.20in} \end{figure}](img27.gif) |
An alternate scheme, also appearing in ref. [29], is to employ a reverse Monte Carlo algorithm. In this approach the atoms forming the polymer backbone are simply moved at random so that all configurations which generate acceptable structure functions are sampled. In this case the Monte Carlo partition is governed by the chi-square error function arising from the differences between the experimental and model-derived structure functions. For polyethylene the best-fit torsion-angle distributions generated by RMC compare very favorably with those obtained from molecular simulations.
While there is considerable merit in all these approaches, continued progress
requires implementation of more tightly nested modeling schemes. For a true
refinement of the experimental data an indirect comparison with molecular
simulation has its limitations. Often there is an incomplete knowledge of the
molecular force-fields, particularly with respect to torsion potentials.
Ideally the potentials used by an appropriate simulation could be modified in an
iterative process using direct and immediate comparisons to the experimental PDF
data. In this way the simulation algorithms could be optimized by integrating
them into a comprehensive refinement scheme. Moreover it is important to
recognize that there still exists an extensive range of polymeric materials
which are not easily mimicked using the current generation of computer
simulations. In particular there is considerable difficulty in obtaining
physically correct solutions for polymers having shallow minima in their
torsional potentials (e.g., the polysilanes), extensive ![]() -conjugation
along the backbone (as in the case of conducting polymers), or polymer hosts having
ionic interactions (as in the of polyelectrolytes). Semi-crystalline
and liquid crystalline polymers are also problematic because of the
intrinsically slow kinetics and the subtle competing interactions which often
exist at the shorter length scales.
-conjugation
along the backbone (as in the case of conducting polymers), or polymer hosts having
ionic interactions (as in the of polyelectrolytes). Semi-crystalline
and liquid crystalline polymers are also problematic because of the
intrinsically slow kinetics and the subtle competing interactions which often
exist at the shorter length scales.
An unrestricted RMC approach may be problematical as well. Since, a priori, no atom interactions are used, unphysical and energetically unfavorable configurations may be extensively sampled and thus leading to a maximal set of potential solutions. In many instances the local correlated motions of nearest-neighbors are well understood. A hybrid scheme in which these known attributes are incorporated into the RMC would then lead to a more tightly constrained set of solutions.
The choice of which data representation to use is also an important
consideration. As is the case for other classes of materials, a combined
real-space and momentum space refinement of the data can be advantageous since
the two representations emphasize different attributes. As noted previous, the
G(r) profiles derived from x-ray scattering often contain unphysical
artifacts at low r. These artifacts appear across the entire S(q) profiles
and complicate refinements which utilize the structure function. By weighting the
refinement towards G(r) data points at larger r's one can minimize their
impact. On the other hand, the intrachain G(r) is necessarily superimposed on
the interchain G(r) and refinement of this data requires a prior knowledge of
the interchain pair correlation function. In contrast this interchain feature
is limited to the low q range of the structure function so that restricting
a refinement to the higher q's guarantees that only intrachain components
are used when optimizing an intrachain model. Thus a carefully orchestrated
refinement of both sets of data can lead to a more rapid convergence towards
a physically appropriate model. In many instances local chain polymers structures
can be found which work well in either r-space or q-space but not both.
A MODEL HOST POLYMER FAMILY
As a model test system, we have recently begun a series of experiments to ascertain
the local intrachain and interchain structure in various polysilane derivatives.
Polysilanes are well-known polymers ([-SiRR'-]n) comprised of only
![]() -bonded silicon atoms along the polymer backbone and short alkyl and/or
alkoxy segments as the side-chains. From a structural perspective there is an
intriguing UV absorption feature[32,33], due to a
-bonded silicon atoms along the polymer backbone and short alkyl and/or
alkoxy segments as the side-chains. From a structural perspective there is an
intriguing UV absorption feature[32,33], due to a ![]() transition along the
Si-atom backbone, which has been shown to be extremely sensitive to the choice of
side chain constituents, sample temperature and processing history. In the
limiting case of an all-trans main-chain conformation, typically seen in some
symmetric alkylsilane samples[34,35] such as poly(di-hexylsilane), the peak in the UV
absorption is centered near 370 nm. Helical and other, at present, unknown chain
conformations exhibit transitions which are ``blue''-shifted to shorter
wavelengths. One long-standing question is simply how small changes in the local
main-chain and side-chain conformations influence the nature of this
transition. A proper interpretation requires a quantitative assessment of the
local main-chain and side-chain structure in a variety of settings. Direct
molecular simulations are of limited use at present because ab initio
calculations of the Si-Si-Si-Si torsion potential obtain only slight energy
differences between trans, gauche and various intermediate
conformations[36]. Hence there is a strong impetus to use direct probes of local
structure to gain further insight.
transition along the
Si-atom backbone, which has been shown to be extremely sensitive to the choice of
side chain constituents, sample temperature and processing history. In the
limiting case of an all-trans main-chain conformation, typically seen in some
symmetric alkylsilane samples[34,35] such as poly(di-hexylsilane), the peak in the UV
absorption is centered near 370 nm. Helical and other, at present, unknown chain
conformations exhibit transitions which are ``blue''-shifted to shorter
wavelengths. One long-standing question is simply how small changes in the local
main-chain and side-chain conformations influence the nature of this
transition. A proper interpretation requires a quantitative assessment of the
local main-chain and side-chain structure in a variety of settings. Direct
molecular simulations are of limited use at present because ab initio
calculations of the Si-Si-Si-Si torsion potential obtain only slight energy
differences between trans, gauche and various intermediate
conformations[36]. Hence there is a strong impetus to use direct probes of local
structure to gain further insight.
From the perspective of technique development, the polysilane polymer family also appears to have a significant advantage over conventional polymeric materials. The presence of an all-silicon backbone significantly enhances the strength of the x-ray scattering signal as compared to lower Z hosts. Thus the PDF profile will be strongly dominated by scattering from the well-defined (CHx-Si-CHx) core units. Moreover the nearest-neighbor Si-Si pair distance of 2.4 Å is well separated from the nominal 1.9 Å and 1.5 Å pair distances of C-Si and C-C, respectively, so that at intermediate length scales (from 2 to 5 Å) the pertinent information is better differentiated. For refinements against the structure function, H(q), the larger repeat unit also guarantees that more relevant scattering features are concentrated in the 20-25 Å-1 q-range now easily accessible with existing light sources and instrumentation.
The smallest poly(alkyl) derivatives exhibiting thermochromism are the symmetric
poly(di-ethylsilane) (PdeS) and the asymmetric poly(methy-n-propylsilane)
(PmpS). At room temperature PdeS is crystalline with an all-trans
Si-backbone conformation while PmpS is found to be semi-crystalline with a
monoclinic approximate having lattice parameters of a=8.38 Å,
b=10.13 Å, c=3.92 Å and ![]() =66
=66![]() where c is along the Si
backbone direction[37]. Nominally an all-trans conformation is consistent with
the Bragg scattering analysis but the UV absorption maximum is near 320 nm which is
much more suggestive of a non-planar arrangement. Slightly longer methyl-
n-alkyl polysilanes exhibit a somewhat different thermal behavior (with
thermochromism occurring at reduced temperatures) and with chain conformations which
are, at present, unknown. To be entirely fair there are some drawbacks.
For instance, existing synthetic routes produce
materials which are atactic so that isotactic and syndiotactic dyads occur at
random and in equal proportions. Modeling of an atactic system is, necessarily,
more complicated than for more regular chain structures. Polysilanes are also
found to be radiation sensitive. Exposure to the x-ray beam for over 24 hours
caused no discernible differences in the scattering profiles despite a certain
drop in the molecular weights.
where c is along the Si
backbone direction[37]. Nominally an all-trans conformation is consistent with
the Bragg scattering analysis but the UV absorption maximum is near 320 nm which is
much more suggestive of a non-planar arrangement. Slightly longer methyl-
n-alkyl polysilanes exhibit a somewhat different thermal behavior (with
thermochromism occurring at reduced temperatures) and with chain conformations which
are, at present, unknown. To be entirely fair there are some drawbacks.
For instance, existing synthetic routes produce
materials which are atactic so that isotactic and syndiotactic dyads occur at
random and in equal proportions. Modeling of an atactic system is, necessarily,
more complicated than for more regular chain structures. Polysilanes are also
found to be radiation sensitive. Exposure to the x-ray beam for over 24 hours
caused no discernible differences in the scattering profiles despite a certain
drop in the molecular weights.
 |
Our present modeling approach employs a direct refinement of the various bond
angles, bond lengths and dihedral angles within a single ``atactic'' eight Si unit
oligomer model having periodic boundary conditions. Hard core packing constraints
prevent overlap of neighboring atoms. This scheme represents an attempt to find a
single conformational setting which adequately reproduces the scattering data. As
such it is quite limited but it is still illustrative for demonstrating the
sensitivity of PDF analysis in studies of local polymer structure. Figure 6
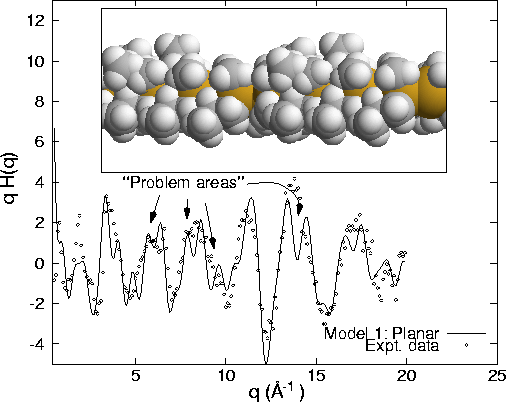
compares a q-weighted H(q) from a room temperature PmpS sample and a model
restricted to a trans-planar configuration with refinement of only the alkyl side
chains. As denoted by the arrows, a fixed planar backbone structure is clearly an
unphysical description of the local chain structure. An equally as poor G(r)
comparison is also obtained (not shown). By allowing for variations in both the
Si-Si-Si bond angles and the Si-Si-Si-Si dihedral angles substantial improvements
are obtained. The current ``best-fit'' model is shown in Fig. 7 using both q-
and r-space representations. On average there appears to be a 20![]() deviation from planarity although it can exceed 40
deviation from planarity although it can exceed 40![]() across at least one of
the eight Si-Si-Si-Si linkages. The presences of these large twists, in
combination with random disorder, is consistent with the shorter wavelength UV
absorption maximum found experimentally. At present there also appears to be
strong cross-correlations between the local Si bond and dihedral angles. With
continued improvements in the modeling algorithms it may be possible to
quantitatively specify this behavior.
across at least one of
the eight Si-Si-Si-Si linkages. The presences of these large twists, in
combination with random disorder, is consistent with the shorter wavelength UV
absorption maximum found experimentally. At present there also appears to be
strong cross-correlations between the local Si bond and dihedral angles. With
continued improvements in the modeling algorithms it may be possible to
quantitatively specify this behavior.
![\begin{figure}
\begin{center}
\begin{center}
\includegraphics [height=4.0in]{fig...
...eps}
\end{center} \vspace*{-.1in}
\end{center} \vspace*{-.150in} \end{figure}](img34.gif) |
![\begin{figure}
\begin{center}
\begin{center}
\includegraphics [height=4.0in]{fig...
...eps}
\end{center} \vspace*{-.1in}
\end{center} \vspace*{-.150in} \end{figure}](img35.gif) |
Finally, in Figure 8, preliminary G(r) (as r [G(r)-1]) profiles are shown for a poly(methyl-n-hexylsilane) sample observed at temperatures clearly above and below the reported thermo-chromic transition point. The actual UV absorption spectra exhibit a clear isosbestic point and are indicative of a distinct two-phase coexistence over the transition region. The two displayed G(r) profiles exhibit a number of differences suggestive of distinct changes in the chain structure. In the 3 to 5 Å region the peaks and valleys appear measurably sharper in the low temperature profile. In addition, pair correlation peaks centered near 5.9 Å and 7.8 Å become more pronounced in the low temperature curve. These two latter features may be tentatively assigned to the Si-Si-Si-Si and Si-Si-Si-Si-Si pair distances and, if this is valid, then imply a transition to a more planar main chain conformation. Once again improvement modeling will be required before a proper assessment of this structural transformation can be reached. Despite the tentative nature of the polysilane data and the various interpretations, these spectra hopefully demonstrate the structural information which can be extracted from PDF analysis of polymer systems.
CONCLUSIONS
Existing polymer PDF studies have, on the whole, only touched upon a few of the possible host systems which could benefit from this methodology. The continuing development of computer modeling algorithms will rapidly advance the quantitative analysis capabilities of the technique. With the ongoing development of dedicated x-ray and neutron spectrometers, an even larger range of structural studies will be available in the future. The high-q ranges now becoming accessible should motivate a reevaluation of the theoretical elastic and Compton scattering profiles. Better knowledge of these attributes will reduce the guess work now required to appropriately scale experimental spectra.
It is important to note that PDF studies of oriented polymer samples are also possible. This would allow the anisotropy in the local structure to be better reconstructed thus yielding less uncertainty in the structures obtained during the refinement process. However this would significantly increase the number of calculations necessary for these refinements.
In addition to the conventional x-ray scattering results briefly reviewed here, there also are an expanding number of polymer PDF studies employing anomalous scattering techniques. These include NiBr-doped polyelectrolytes[38,39], HBr-doped polyaniline[40] and, most recently, Li6 and Li7 substitutions in polyethylene oxide[41]. These studies are potentially more powerful than the much more commonly used EXAFS technique because anomalous PDF methods can resolve structural features beyond 5 Å and, in the case of neutron scattering, investigate a large number of low Z materials.
There are many avenues for the application of PDF techniques in the study of local structure in polymeric materials.
ACKNOWLEDGMENTS
This support of this work by a NSF grant (DMR-9631575, M.J.W.) is gratefully acknowledged. I also wish to acknowledge fruitful discussions and collaborations with G.R. Mitchell, B.R. Mattes, Robert West and J.R. Koe.